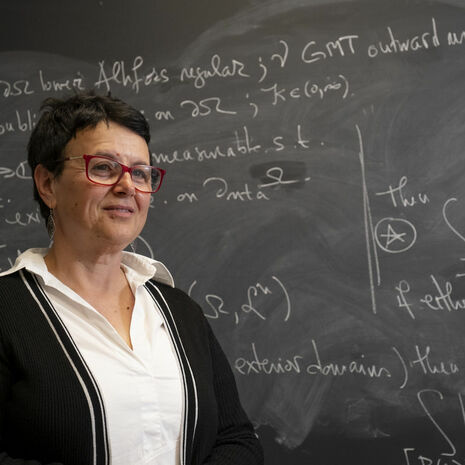Tarantulas bend rules to continue scampering after losing two legs Tarantulas bend rules to continue scampering after losing two legs by Kathryn Knight June 19 [field_tu_pg_body_text]
Class of 2025: Livestreams, speakers, programs and more for both the undergraduate and graduate student ceremonies Class of 2025: Livestreams, speakers, programs and more for both the undergraduate and graduate student ceremonies by Greg Fornia May 05 [field_tu_pg_body_text]
Temple celebrates a historic trio of Goldwater Scholars in 2025 Temple celebrates a historic trio of Goldwater Scholars in 2025 April 30 [field_tu_pg_body_text]
CIS/ACM Award Winners 2025 CIS/ACM Award Winners 2025 by Greg Fornia April 25 [field_tu_pg_body_text]
Math Major D. Palomino receives Goldwater Scholarship Math Major D. Palomino receives Goldwater Scholarship by Mathematics April 23 [field_tu_pg_body_text]
New Edition of Mitrea’s Hodge-Laplacian Research Monograph New Edition of Mitrea’s Hodge-Laplacian Research Monograph by Mathematics April 23 [field_tu_pg_body_text]
Mathematics's Charles Osborne, CST ’10, recipient of Temple's Lindback Distinguished Teaching Award, on memorable moments, proudest accomplishments and inspiration for teaching Mathematics's Charles Osborne, CST ’10, recipient of Temple's Lindback Distinguished Teaching Award, on memorable moments, proudest accomplishments and inspiration for teaching by Greg Fornia April 16 [field_tu_pg_body_text]
Dennis Terry, Associate Professor of Earth and Environmental Science, leads a field trip at a recent Geological Society of America Meeting Dennis Terry, Associate Professor of Earth and Environmental Science, leads a field trip at a recent Geological Society of America Meeting by Atsuhiro Muto April 16 [field_tu_pg_body_text]
Frank Spano, professor of chemistry, earns Temple University Faculty Research Award Frank Spano, professor of chemistry, earns Temple University Faculty Research Award by Greg Fornia April 16 [field_tu_pg_body_text]
30 Under 30: Binh Le, CST '17, biology major and surgical podiatrist who dream of becoming a superhero 30 Under 30: Binh Le, CST '17, biology major and surgical podiatrist who dream of becoming a superhero by Greg Fornia April 04 [field_tu_pg_body_text]
Congrats to Benjamin Prather, Recipient of ACS Student Travel Award Congrats to Benjamin Prather, Recipient of ACS Student Travel Award by Vladi Wilent March 28 [field_tu_pg_body_text]
May 1: Franklin Institute Awards Symposium: Density Functional Theory honoring John Perdew May 1: Franklin Institute Awards Symposium: Density Functional Theory honoring John Perdew by Greg Fornia March 25 [field_tu_pg_body_text]
30 Under 30: Haley Lockstein, CST '19, biology graduate honored by Temple as an 'innovator' 30 Under 30: Haley Lockstein, CST '19, biology graduate honored by Temple as an 'innovator' by Greg Fornia March 20 [field_tu_pg_body_text]
Livestream: Searching for new species in the South Sandwich Islands with Biology Department chair Erik Cordes Livestream: Searching for new species in the South Sandwich Islands with Biology Department chair Erik Cordes by Greg Fornia March 19 [field_tu_pg_body_text]
Spring forward? How climate change is rushing the spring season Spring forward? How climate change is rushing the spring season by Jonny Hart March 18 [field_tu_pg_body_text]
Rebecca Feldman, CST '18, EES graduate one of Temple's 30 Under 30 Rebecca Feldman, CST '18, EES graduate one of Temple's 30 Under 30 by Greg Fornia March 17 [field_tu_pg_body_text]
Students in the Philadelphia region shine at Temple’s annual science fair Students in the Philadelphia region shine at Temple’s annual science fair March 12 [field_tu_pg_body_text]
Alex Cagle, CST ’18: EES graduate chosen as part of Temple's 2025 30 Under 30 Honorees Alex Cagle, CST ’18: EES graduate chosen as part of Temple's 2025 30 Under 30 Honorees by Greg Fornia March 12 [field_tu_pg_body_text]
Charles Osborne receives Lindback Award Charles Osborne receives Lindback Award by Mathematics March 11 [field_tu_pg_body_text]
Erik Cordes on the discovery of ocean life at the bottom of the Southern Ocean Erik Cordes on the discovery of ocean life at the bottom of the Southern Ocean by JayDiii Grattepanche March 03 [field_tu_pg_body_text]