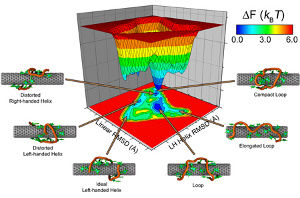
Misdirected protein phosphorylation is frequently associated with human diseases, particularly cancer, neurological disorders, HIV, and diabetes. Over the last decade, there has been a growing interest in applying protein kinase inhibitors in cancer therapy. The first generation of protein kinase inhibitors use a traditional approach with a small drug molecule blocking the active site and is primarily targeting the cyclin dependend kinase 9 (CDK-9) activity. CDKs and their cyclin partners form a heterodimer, in which the CDK contains the enzymatic domain and the cyclin is the regulatory domain.
The goal of the project is to find more selective and specific ways to control CDK activity by regulating protein-protein binding instead. To that effect, novel drugs have to be designed on the basis of binding maps computed from all-atom and coarse grain molecular dynamics simulations.
References:
- V. Caracciolo, G. Laurenti, G. Romano, V. Carnevale, A. M. Cimini, C. Crozier-Fitzgerald, E. Gentile Warschauer, G. Russo, A. Giordano, Cell Cycle, 11, 1202-1216, (2012).
- G. Romano and A. Giordano, Cell Cycle, 7, 3664-3668, (2008).
Two-component Systems of Virulent Bacteria
Pathogenic bacteria contain two-component systems (TCS) that sense the environment within a host and activate mechanisms related to antimicrobial resistance.
Unraveling how binding of their ligands on the outside of the cell transmits a signal to the inside would help to understand the mechanisms of signal transduction.
This work is done in collaboration with Prof. William F. DeGrado (UCSF)
References:
- D. S. Goldberg, C. S. Soto, C. D. Waldburger, W. F. DeGrado., J. Mol. Biol., 379, 656-665 (2008).
- A. M. Stock, V. l. Robinson, P. N. Goudreau, Annu. Rev. Biochem., 69, 183-215 (2000).
Unraveling the Enzymatic Mechanism of the Protein Farnesyltransferase
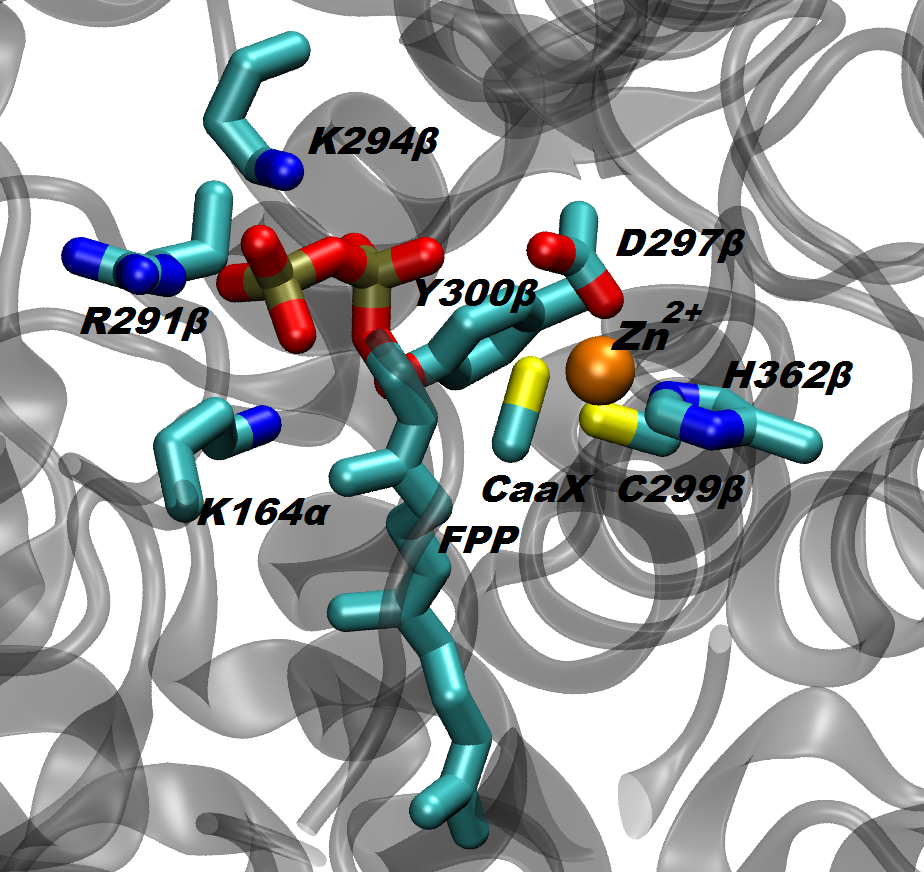
The protein farnesyltransferase (FTase), a Zn-metalloenzyme, catalyzes the transfer of the 15-carbon farnesyl group from the farnesyl diphosphate (FPP) to acceptor proteins that contain the so-called “CaaX” motif at the C-terminus. FTase activity is crucial in signal transduction pathways such as proliferation and apoptosis of cells. In fact, the Ras superfamily and small GTPases including Ras, Rho and Rab, are important examples of proteins that are activated by FTase function. Nowadays, FTase represents one of the promising targets for anti-cancer drug design, being involved in the activation of oncogene proteins such as mutated Ras, which are related to the development of ~20-30% of human cancers.
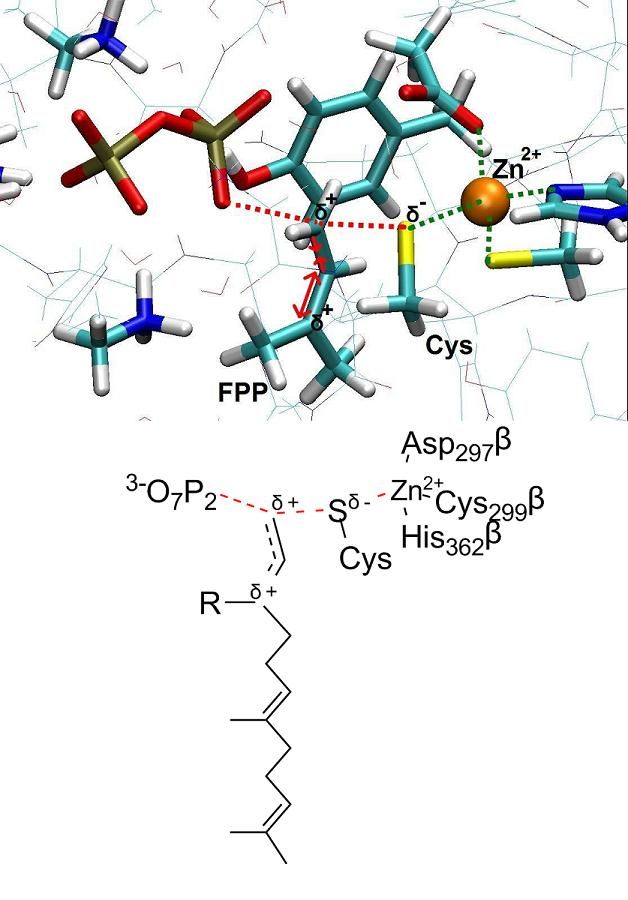
Several distinct hypotheses have been proposed based on various experimental findings in regard of the chemical reaction. A detailed picture of the reaction mechanism and its transition state would certainly improve our understanding of FTase enzymatic activity. Towards this aim, we focus on the chemical step of the catalytic cycle and present a theoretical investigation of the farnesylation mechanism. Our computations employ classical molecular dynamics (MD) and ab initio Car-Parrinello QM/MM calculations. Overall, our finding indicates that the reaction mechanism is an associative mechanism with dissociated character, which fits the experimental data well.
References:
- M.-H. Ho, M. De Vivo, M. Dal Peraro, and M. L. Klein, J. Chem. Theory Comput., 5, 1657 (2009)
Study of Redox Reaction in Superoxide Reductase through a DFT + Hubbard U Approach
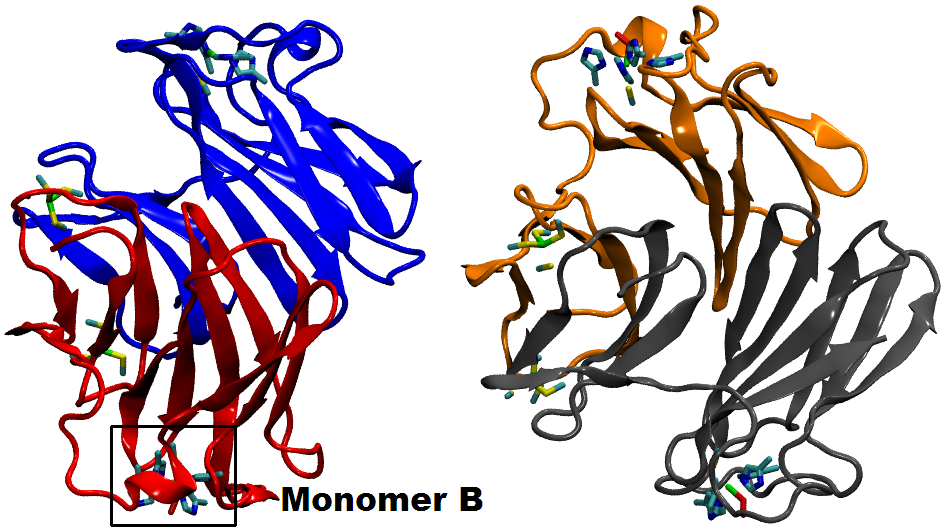
Many biological processes (including respiration, protein cleavage and toxic particles removal) are catalyzed by transition-metal containing enzymes. While localized d-states of the transition-metal ions are central in these catalytic reactions, accurate descriptions of these states face great challenges when using DFT method. In this work, we adopt a QM/MM technique coupled with a DFT + Hubbard U scheme in order to study the transition metal enzyme called Superoxide Reductase (SOR), with an appropriate description of the d-state electrons.
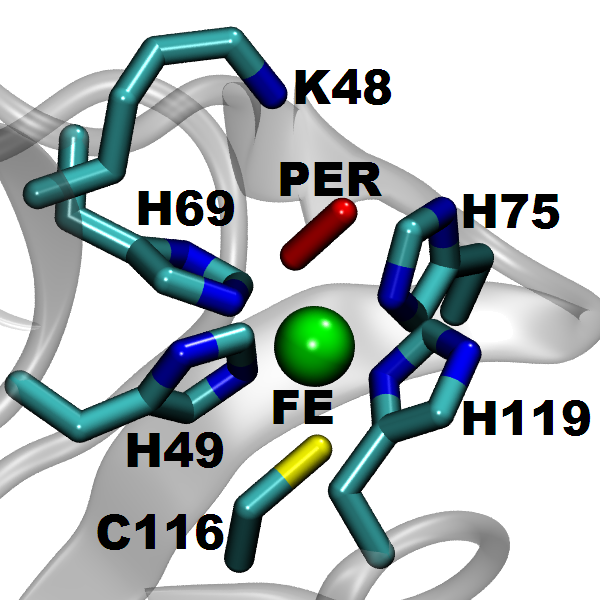
SOR (see the protein and active site structure shown in the figure), found in several anaerobic bacteria, is important in toxic oxygen derivatives removal, by catalyzing the one-electron reduction of superoxide to hydrogen peroxide. It prevents the production of molecular oxygen and opens a new way for the therapeutic treatment of diseases related to the presence of superoxide, through the ideation of suitable SOR mimics. We study the structure and energetics of the iron-dioxygen intermediates at different stages of the reduction process. According to our results, the introduction of the Hubbard U correction leads to an improved description of the intermediate structures and the reaction mechanism. Finite temperature studies with better descriptions of structures and energetics allow us to obtain a reliable quantitative analysis of the enzymatic reaction rate and mechanism.
References:
- P. H.-L. Sit, A. Migliore, M.-H. Ho and M. L. Klein, J. Chem. Theory Comput., 6, 2896-2909 (2010)
Electron Transfer Problems in Aqueous Environments
Electron-transfer reactions are ubiquitous processes in organic and inorganic redox reactions. In the diabatic limit, the transfer integral is small, and the system has to diffuse up to the intersection several times before an electron can tunnel. The reaction rate for such a weak coupling case can be written as
where Hif is the transfer integral between the initial and final states. ρFC is the density of states weighted by the Franck-Condon factor and thermally averaged. This term becomes
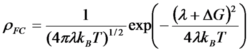
in the classical case. In the formula, λ is the reorganization energy and ΔG is the energy of the reaction.
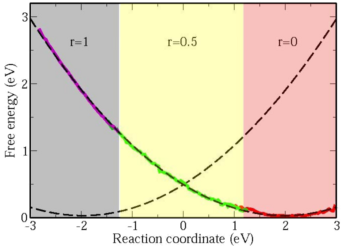
To obtain quantitative descriptions of electron-transfer reactions fully from first-principles, we developed a novel method to calculate free-energy surfaces of electron-transfer reactions from ab initio molecular dynamics. Sampling of phase space is done with a penalty functional added to the standard density functional so that the oxidation states of ions can be controlled. This penalty functional opens up the possibility of performing out of ground state DFT calculations, and is also a practical way to correct for the self-interaction in DFT.
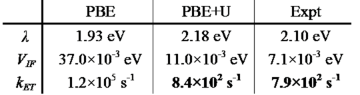
With this penalty functional technique, we calculated the free-energy surfaces of the self-exchange reaction between aqueous ferrous and ferric ions fully from first-princieples (see figure). We found that the free-energy curves are parabolic, validating the linear solvation model first proposed by Marcus. The reorganization energy obtained is 1.93 eV, which is in good agreement with the experimental value of 2.1 eV, and a big improvement over classical and semi-classical calculations in quantitatively describing electron-transfer reactions. A further improved value of 2.18 eV is obtained by including Hubbard U correction to accurately describe the strongly localized d-electrons.
We then calculated the transfer integral by adopting a recently developed ab initio method. Combining the reorganization energy and the transfer integral, we can estimate the ET reaction rate constant fully ab initio. The result of 8.4 × 102 s-1 is in excellent agreement with the experimental estimate of 7.9 × 102 s-1.
References:
- A. Migliore, P. H.-L. Sit, and M. L. Klein., J. Chem. Theory Comput., 5, 307 (2009)
- P. H.-L. Sit, M. Cococcioni, and N. Marzari. J. Electroanal. Chem., 607, 107 (2007)
- P. H.-L. Sit, M. Cococcioni, and N. Marzari. Phys. Rev. Lett., 97, 028303 (2006)
Molecular Simulation of Polymer Self-Assemblies and Mixtures
A worm-like micelle is a classic example of a molecular self-assembly that possesses a unique elongated shape. The polymer hydrophobic tails self assemble in the core of the micelle, while the hydrophilic groups maximize contacts with water by self-assembling in the corona of the micelle. Using mesoscopic simulation techniques, dissipative particle dynamics (DPD), we can test the stability of worm-like micelles at different hydrophilic block fractions. Worm-like micelles exist in a narrow region of the phase diagram. If the hydrophobic portion of the diblock copolymer is degradable, such as PCL (poly(caprolactone) (PCL) or poly(lactic acid) (PLA), this leads to shortening of the relative molecular weight of the tail, and an increase in the polydispersity, over time. The leads to instabilities, and ultimately, a breakup of the worm-like micelle morphology. We emulate this process in simulation by mixing in diblocks of differing hydrophobic length. At a critical concentration, this leads to a breakup of the previously stable worm. This instability begins with radial undulations along the core, bud formation at the end of the micelle, and finally expulsion of a spherical micelle from the end-cap. Analysis of the mean curvature of the worm micelle during break-up proves a simple linear relationship with the mean interfacial concentration of each component diblock copolymer relative to each equilibrium unmixed micellar phase.

Other examples of self-assembly structures are bilayers. We use Coarse-Grained Molecular Dynamics (CGMD) to gain insight about the strong lateral segregation observed in polymersomes composed by neutral and charged diblock-copolymers (Christian et al, 2009) in the presence of calcium. Our approximation involved toy models in which the charged groups were represented with LJ interaction sites. These simple models are able to successfully reproduce the morphology phase diagram for charged diblock copolymers as well as the spontaneous segregation observed in bilayers composed by charged and neutral copolymers. The shape of the aggregates appeared correlated to its state: crystalline patches are irregular, liquids are regular. As suggested by experiments, after several nanoseconds of simulation, we found strong correlation between the local composition of the two leaflets (registration). Simulations suggest that differentiated interlealet interaction between ordered and disordered phases would contribute to the registration.
References:
- S. M. Loverde, M. L. Klein and D. E. Discher, Advanced Materials, 24, 3823-3830 (2012)
- D. A. Pantano, P. B. Moore, M. L. Klein and D. E. Discher, Soft Matter, 7, 8182-8191 (2011)
- S. M. Loverde, V. Ortiz, R. D. Kamien, M. L. Klein and D. E. Discher, Soft Matter, 6, 1419-1425 (2010)
- D. A. Christian, A. Tian, W. G. Ellenbroek, I. Levental, K. Rajagopal, P. A. Janmey, A. J. Liu, T. Baumgart & D. E. Discher, Nature Materials 8, 843-849 (2009)
- Y. Geng and D. E. Discher.Journal of the American Chemical Society, 127, 12780-12781 (2005)
Self-assembly of Surfactants using Implicit Solvent Models
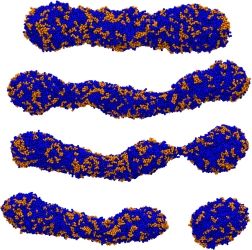
Surfactants are used in many industrial applications, such as detergents in cleaning products, as visco-elastic drilling fluids, or for solubilization of lipid molecules. Above the so-called critical micelle concentration (cmc), surfactants self-assemble into aggregates of characteristic morphologies (spherical or worm-like micelles, bilayers) depending, e.g., on the chemical architecture of the surfactant or the composition of the surfactant solution. It is computationally very challenging to model the self-assembly process of such systems, due to the required systems sizes and the long aggregation time (>1μs). Coarse grain simulations and implicit solvent models allow studies of self-assembly processes on the required time and length scales. A recently developed implicit solvent model of ionic surfactants is extended to study the effect of divalent salt ions (Ca2+ or Mg2+) on the micellization properties. Furthermore we investigate micellar properties when different surfactant types are mixed, such as sodium dodecyl sulfate (SDS) with polyethylene glycol (PEG) surfactants. It has been experimentally observed that the composition of the surfactant mixture and length of the PEG head group have significant impact on the morphologies of the micelles, e.g., the formation of spherical and worm-like micelles. We are particularly interested in such synergetic effects, and the ability to quantitatively predict the cmc and agggegate structures which is not feasible using conventional atomistic simulation approaches. We combine Grand Canonical Monte Carlo and atomistic Molecular Dynamics simulations to parameterize the model and to study the mentioned self-assembly properties. The image shows a simulation snapshot of an ellipsoidal micelle composed of a mixture of sodium dodecyl sulfate [SDS; grey spheres attached to light blue chain] and dodecyl polyethylene glycol surfactants [red and light blue chain molecules] in presence of NaCl salt [small dark blue and green particles].
References:
- A. Jusufi et al., J. Phys. Chem. B 112, 13783 (2008)
Hybrid Nanomaterials
Meeting the demands of the rapidly advancing nanotechnological frontier requires novel, multifunctional nanoscale materials. Among the most promising nanomaterials to fulfill this need are biopolymer-carbon nanotube hybrids (Bio-CNT). Bio-CNT consists of a single-walled carbon nanotube (CNT) coated with a self-assembled layer of biopolymers such as DNA or protein. Experiments have demonstrated that these nanomaterials possess a wide range of technologically useful properties with applications in nanoelectronics, medicine, homeland security, environmental safety and microbiology. We use all-atom molecular dynamics (MD), parallel tempering replica exchange (REMD) and free energy methods gain insight into properties and behavior of these unique nanomaterials.
Simulation of Nano-Biosensors: Carbon Nanotube Functionalized with the Coxsackie-Adenovirus Receptor
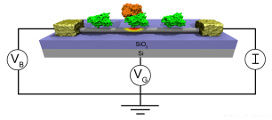
Detection of viruses and other harmful biological agents has important applications in homeland security and medicine. Recently, the experimental nanoscience group of Dr. Charlie Johnson at the University of Pennsylvania has developed sensors capable of detecting proteins associated with the adenovirus (one of the viruses responsible for the common cold). These sensors are depicted below. They consist of the Coxsackie-adenovirus receptor protein (green) attached to a carbon nanotube (gray cylinder). Knob proteins (orange) that are located on the surface of the adenovirus bind to this receptor protein. This binding event changes the electrical properties of the carbon nanotube. Thus, by monitoring the electrical properties of the carbon nanotube, one can detect the presence of the adenovirus. ICMS uses a variety of computational techniques to help rationalize the fabrication and operation of these sensing devices.
References
- R.R. Johnson, A.T.C. Johnson, M. L. Klein, Small, 6, 31, (2010).
- R.R. Johnson, B.J. Rego, A.T.C. Johnson, M.L. Klein, J. Phys. Chem. B, 113, 11589 (2009)
- R.R. Johnson, A. Kohlmeyer, A.T.C. Johnson, M. L. Klein. Nano Lett., 9, 537 (2009)
Aqueous Ionic Liquid Solutions
Ionic liquids made up of organic cations and organic or inorganic anions have very low vapor pressure, which sets them apart from commonly used organic solvents. This makes them attractive for use as solvents for synthesis and catalysis and other industrial applications.
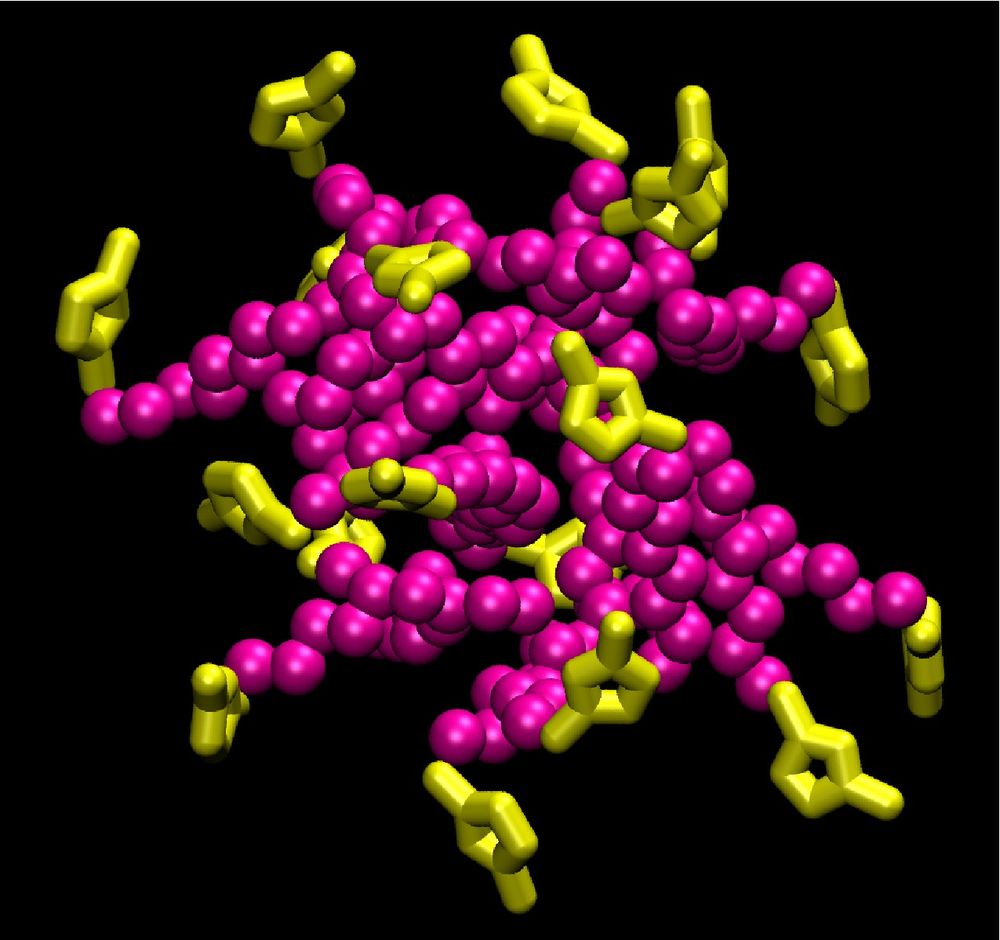
Atomistic molecular dynamics (MD) studies have been performed to study aggregation in these systems. Aqueous solutions of [C10mim][Br] spontaneously form cation aggregates. Simulations of the vapor-liquid interface of the same system shows that the alkyl tails of the cations aggregate at the surface to minimize the unfavorable interactions with water. In higher concentration, the cations also form aggregates in the aqueous phase with polar head groups (imidazolium ring) pointing outwards and the alkyl tails inwards. The determined aggregate sizes agree with experimental observations. The simulated systems appear to be in a metastable state with reference to the aggregation numbers. To extend the accessible observation times, a coarse grained model has been developed for this system.
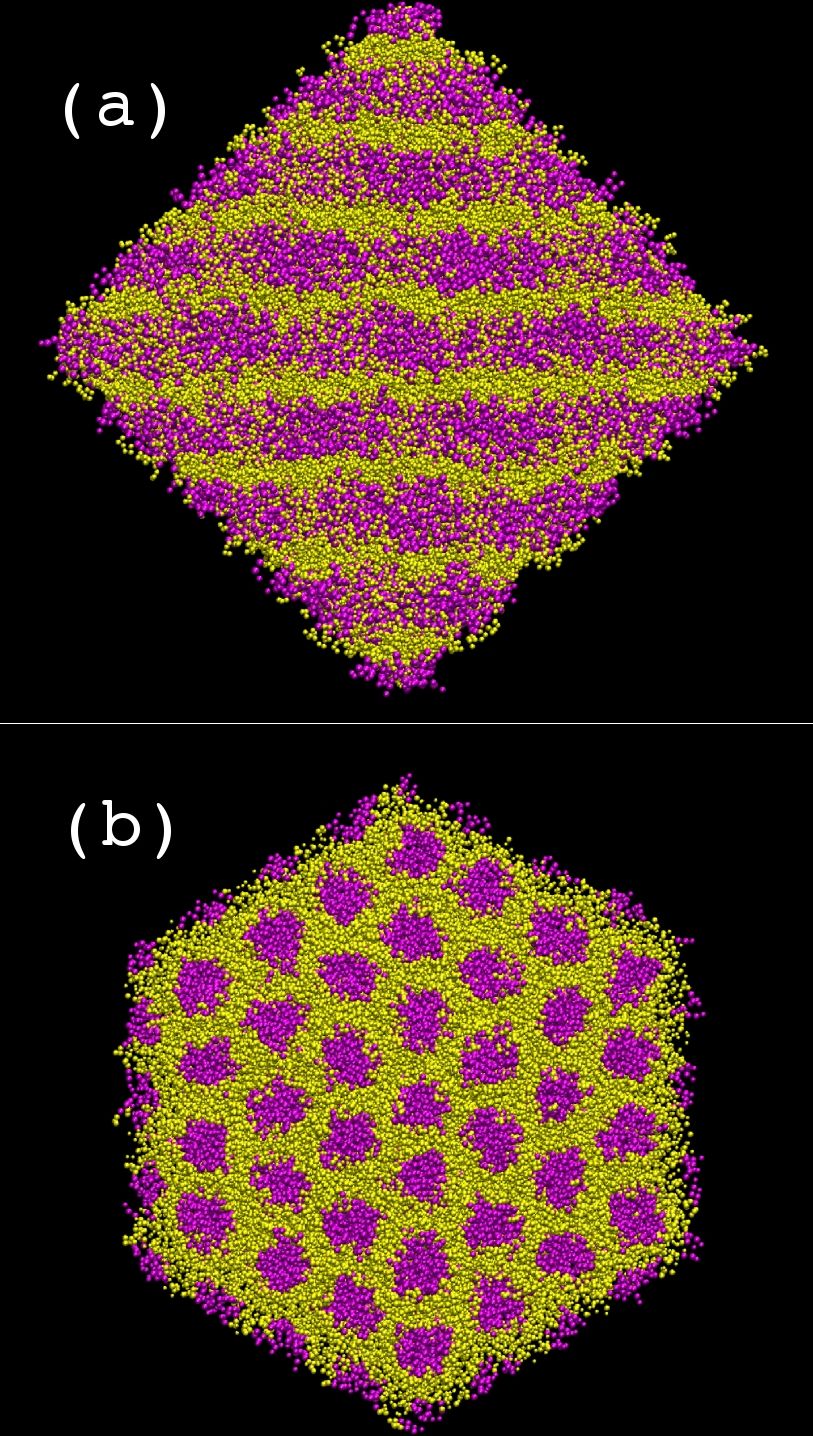
Coarse grain MD simulations of aqueous [C10mim][Br] solutions at several concentrations provide insight into their microscopic structure. At 37% (w/w) water concentration there is a separation of hydrophilic (head groups, anions and water, yellow) and hydrophobic regions (tail groups, magenta) forming a hexagonal columnar phase (see figure on the right: (a) side view of columns, (b) top view of columns).
In dilute aqueous solutions of [C10mim][Br] quasi-spherical micelles form spontaneously. Initially, a large number of monomers and few small aggregates are observed. Yet over time the aggregates grow in size by incorporating those monomers.
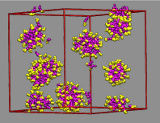
For the small aggregates it is difficult to grow by merging with other micelles due to repulsion of their charged surfaces. The observed aggregates are poly-disperse and typically contain between 40 and 60 cations. The bulk region of the vapor/liquid interface of aqueous ionic liquids shows similar aggregation behavior.
References:
- B.L. Bhargava, M. L. Klein, J. Phys. Chem. A., 113, 1888 (2009)
- B.L. Bhargava, M. L. Klein, Mol. Phys., 107, 393 (2009)
- B.L. Bhargava, M. L. Klein, Soft Matter, 5, 3475 (2009)
- B.L. Bhargava, M. L. Klein, J. Phys. Chem. B, 113, 9499 (2009)